具体信息
补体蛋白调节特异性抗体的作用,有助于处理和去除免疫复合物,并调节 T 细胞和 B 细胞反应。
补体缺乏症可为遗传病,或由感染导致(例如,复发性脑膜炎球菌或播散性淋球菌感染)或与慢性风湿病或自身免疫性疾病(例如,系统性红斑狼疮或冷球蛋白血症)共病。
诊断基于临床和/或家族史以及特征性血液学检测;只有少数专业实验室提供全面的诊断。
可疑的表现包括 5 岁以上个体患流行性脑脊髓膜炎、复发性细菌感染、无荨麻疹的血管性水肿、肾脏和眼系统出现炎性疾病以及自身免疫性临床表现。
在宿主微生物防御中,补体的作用至关重要。它对特异性抗体的作用进行补充,并且对处理和去除免疫复合物起到辅助作用。[1][2][3] 它还利用在各种免疫细胞上发现的特异性受体来调节适应性免疫应答,从而调节 T 细胞和 B 细胞的应答。补体系统参与造血、脂质代谢、生殖和组织再生。[3] 由于其潜在发炎几率很高,因此需要多种调节蛋白来预防潜在的组织损伤。
如果补体系统过度活化,在发生严重创伤、烧伤或感染后可能会引起多种炎症和致命性疾病,包括脓毒症、全身炎症反应综合征、急性呼吸窘迫综合征,以及多器官功能衰竭。[4] 它与严重的肾病以及神经系统变性疾病有关,如阿尔茨海默病、吉兰-巴雷综合征和多发性硬化。
出现疾病状态伴随补体蛋白缺陷(这能导致侵袭性细菌感染的风险升高,且可能与自身免疫疾病有关),或控制、聚集以及限制补体活性的因子缺陷。[5][6][7][8] 补体缺陷可为先天性,也可为后天获得性(继发于补体消耗性疾病状态)。
据报道除血清羧肽酶 N 之外的所有补体蛋白存在完全缺陷。继发性缺乏由补体消耗引起,而补体消耗则是由炎症、自身抗体(例如,C3 肾病因子;针对 C1q、C1-抑制因子 [C1-INH-Abs] 或因子 H 的自身抗体)、合成减少和/或分解代谢增加或蛋白质丢失综合征导致。
补体系统过度活化也被认为是再灌注损伤的显著效应机理。器官功能障碍可能是由血液透析和体外循环中的人造表面引发的炎性反应引起的。在这些病例中,这可能导致短暂的中性粒细胞减少、肺血管白细胞淤滞,以及更为罕见的过敏性休克。
补体缺乏症状态和相关感染
组分
C1q:类系统性红斑狼疮 (SLE)(>90%,临床上多为重症)、感染
C1r/s(多为共病):类 SLE、类风湿性关节炎 (RA)、感染
C4 (C4A、C4B):类 SLE、感染;纯合:临床上表现为重症;杂合:临床上通常不明显
C2:类 SLE、RA、感染(肺炎)、血管炎,临床上通常不明显
C3:化脓性感染
C5:脑膜炎(奈瑟菌属)、SLE
C6:脑膜炎(奈瑟菌属)、SLE
C7:脑膜炎(奈瑟菌属)、SLE
C8 α-γ/C8 β:脑膜炎(奈瑟菌属)、SLE
C9:奈瑟菌属感染(多为无症状)
因子 B:奈瑟菌属感染
因子 D:奈瑟菌属感染
甘露糖结合凝集素 (MBL):细菌感染(多为无症状)
纤维胶凝蛋白 3 (ficolin 3/H-ficolin):呼吸道感染、坏死性小肠结肠炎
MBL 相关的丝氨酸蛋白酶-2 (MASP-2):呼吸道感染。
调节因子
C1-抑制因子:遗传性血管神经性水肿
C4-结合蛋白
备解素:脑膜炎(奈瑟菌属)
因子 H:感染、非典型溶血尿毒综合征 (aHUS)/C3 肾小球疾病 (C3G);aHUS、C3G/膜增殖性肾小球肾炎 (MPGN)
H 因子相关蛋白 1 (FHR1) (FHR3):aHUS、RA、SLE 常与抗因子 H 自身抗体相关 = 补体因子 H 相关蛋白 (CFHR) 缺乏和补体因子 H (CFH) 自身抗体呈阳性 - HUS
因子·I:感染(脓毒症、脑膜炎、肺炎)、aHUS
CD46/膜辅助蛋白 (MCP):aHUS
CD55/促衰变因子 (DAF):严重的肠病;阵发性睡眠性血红蛋白尿症(PIGA 基因的体细胞突变)
CD59:吉兰-巴雷综合征类似症状、溶血;阵发性睡眠性血红蛋白尿症(PIGA 基因的体细胞突变)。
受体
CR3 (CD18/CD11b):白细胞粘附缺陷
CR4 (CD18/CD11c, LFA-1):白细胞粘附缺陷。
补体蛋白以非活化形式存在于血浆中。补体蛋白总共有 50 多种,包括级联系统的各个组分、调节因子(其中多数受细胞表面限制)和受体。
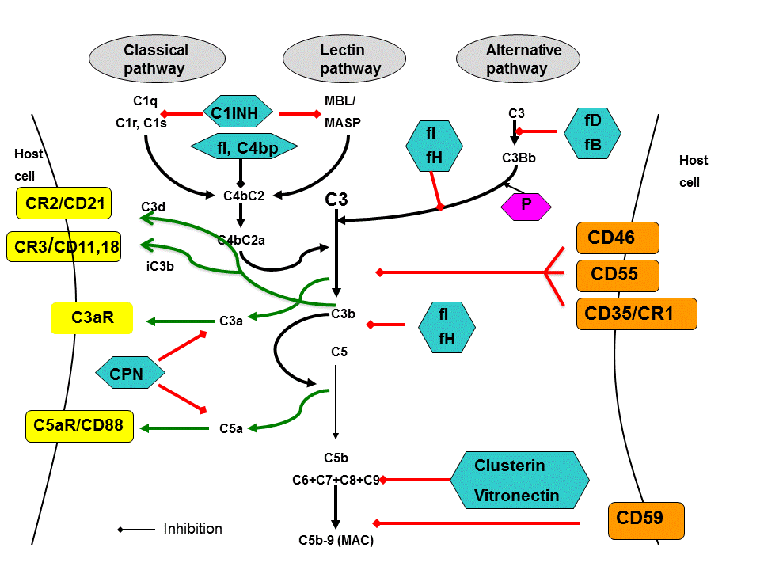 [Figure caption and citation for the preceding image starts]: 补体级联反应的活化和调节。补体通过三种途径被活化,导致以下结果:(1) 产生多种促炎肽(绿色箭头),它们与多种免疫细胞上的特异性受体(黄色)结合;(2) 组装膜攻击复合物 C5b-9 (MAC)。发生各级级联反应时,补体都受可溶性抑制因子(绿松石色)和膜相关抑制因子(橙色)的严格调节。来自 Michael Kirschfink DVM 博士;经获准使用 [Citation ends].
[Figure caption and citation for the preceding image starts]: 补体级联反应的活化和调节。补体通过三种途径被活化,导致以下结果:(1) 产生多种促炎肽(绿色箭头),它们与多种免疫细胞上的特异性受体(黄色)结合;(2) 组装膜攻击复合物 C5b-9 (MAC)。发生各级级联反应时,补体都受可溶性抑制因子(绿松石色)和膜相关抑制因子(橙色)的严格调节。来自 Michael Kirschfink DVM 博士;经获准使用 [Citation ends].
大多数补体蛋白由肝脏分泌,形成急性期应答的一部分。其他组织也能产生补体蛋白,如 D 因子(降脂蛋白)的脂肪细胞。某些补体组分是蛋白酶类,活化状态下在级联酶促反应中裂解下一环节的补体蛋白。扩增步骤的顺序与凝血级联反应相似。
补体级联反应可被 3 条主路线的任一条活化:经典途径 (CP),旁路途径 (AP) 和凝集素途径 (LP)。[1] CP 是特异性抗体应答的关键效应功能,而 AP 和 LP 作为先天免疫系统的一部分,在一线不依赖抗体的细菌感染防御中很重要。补充系统组分的术语与它们被发现的顺序有关,这可解释级联反应的顺序不是按照数字逻辑顺序来安排的原因。AP 的组分蛋白质和调节因子被称为因子(例如,因子 B、因子 H)。
CP 活化主要由抗体(免疫球蛋白)衍生的机制引发。例如,当抗体结合具有免疫复合物或冷球蛋白的特异性抗原时,IgM 和 IgG 抗体(非 IgA)的片段可结晶 (Fc) 区域会在结构上发生改变。当 C1 通过其 C1q 部分与 Fc 区域结合时,CP 将被活化。
在没有抗体的情况下,靶向 C 反应蛋白 (CRP) 也能结合 C1q 并活化 CP。LP 通过结合纤维胶凝蛋白和甘露糖结合凝集素 (MBL) 启动,MBL 是一种众所周知的调理素和一种急性期反应体,结构与 C1q 以及与病原菌和改变的组织上的碳水化合物残基相似。MBL-碳水化合物结合物与 C1 复合物类似,会活化与 MBL 相关的丝氨酸蛋白酶 (MASP),这些酶与 C1 一样能够裂解 C4 和 C2,从而将 LP 连接到 CP。与 CP 不同,AP 主要是通过非抗体(非免疫球蛋白)机制被活化。C3 (C3[H₂O]) 的永久低级水解(在结合因子 B 后,被因子 D 裂解)将产生由备解素稳定的液相 C3 转化酶 (C3b[H₂O]Bb)。在健康状态下,此活动具有自限性;然而,如果新裂解的 C3 与病原菌或变化的组织结合,则 AP 应答将被放大。靶细胞的调节潜力决定了 C3 转化酶是否会在表面上形成、是否发生调理作用,并是否继续级联反应。
一旦补体被任一途径活化,就会产生酶复合物(C3 转化酶),将 C3 裂解成 2 个片段(C3a 和 C3b)。C3a 是较小的片段。C3a 与后来生成的 C5a 类似,是一个促炎症信号分子(过敏毒素)。过敏毒素是趋化因子。他们召集并活化多种炎症细胞,包括中性粒细胞和肥大细胞。C3b 及其在吞噬细胞上的代谢产物 iC3b 的受体可以去除调理的靶点。潜在的病理性免疫复合物(含有与病毒、细菌或自身抗原复合的抗体)会活化 CP 和 C3b,这会标记它们,以便携带 C3b 受体的红细胞通过循环将其清除,并通过网状内皮系统中的吞噬细胞将其清理。在 C3b 与 C3 转化酶结合后,会生成 C5 转化酶,将 C5 裂解为 C5a 和 C5b。C5b 与 C6、C7、C8 和 C9 一起产生亲脂性的膜攻击复合物 (MAC) C5b-9,通过细胞膜裂解引起靶细胞死亡。
需要多种调节蛋白来确保预防或至少限制潜在的补体介导的组织损伤。参与调节和抑制补体系统的其他因子是调节 AP 的因子 H 和因子 I,以及控制 CP 和 LP 的 C1-抑制因子 (C1-INH) 和 C4 结合蛋白 (C4bp)。
C3 转化酶先天不稳定,半衰期短,因此有助于限制和控制补体活化。MCP/CD46 和 DAF/CD55 控制细胞表面上 C3 的活化。可溶性蛋白(凝聚素、玻连蛋白)和 MAC 抑制蛋白 (CD59) 可阻止过量的 MAC 介导的补体裂解。
以下是一些遗传补体缺乏症:
经典途径:C1q、C2、C4 缺乏
旁路途径:因子 B、因子 D 缺乏
凝集素途径:MBL、MASP2 缺乏
C3 缺乏
C5、C6、C7、C8、C9 缺乏
调节因子:C1-抑制因子、因子 H、因子 I、备解素、CD46、CD55、CD59 缺乏。
补体缺乏症约占所有原发性免疫缺陷的 5%,[9] 但可高达 10%。[10] 计算得出遗传性补体缺乏症的患病率约为 0.03%,其中不包括甘露糖结合凝集素 (MBL) 缺乏(估计约占普通白种人群的 5%)。[10] 最常见的补体缺乏症影响 C2 和 MBL,通常无临床症状。遗传性血管水肿(Quincke 水肿)伴 C1-抑制因子缺乏症 (C1-INH-HAE) 的发病率预计为 1/10,000-1/50,000。[11] 但是,补体蛋白缺乏症在患有特定疾病的人中更为常见。在系统性红斑狼疮患者中,有 30% 的患者已患有补体缺乏症[12][13][14] 而在感染了播散型奈瑟菌属的个体中,这一比例约占 20% 左右。随着原发性免疫缺陷分析和补体诊断的改进,预计会有更高的患病率。
既往病史可能揭示出补体缺乏的病因或并发症。获得性补体缺乏症的最常见病因是系统性红斑狼疮 (SLE)。然而,很少有 SLE 患者表现为复发细菌感染,而且,出现复发性细菌感染的原因很多,包括和使用大剂量免疫抑制剂直接相关。
通常会导致消耗的强补体活化常见于患有严重感染、自身免疫性疾病、血管炎、阵发性睡眠性血红蛋白尿症、各种肾病、冷球蛋白血症和部分脂肪代谢障碍的患者。
由补体缺乏症引起的感染可能包括:
复发性化脓感染(如深部脓肿、骨髓炎、肺炎)
菌血症
复发性脑膜炎球菌感染
播散性淋球菌感染。
奈瑟菌属细菌(脑膜炎球菌以及淋球菌)对补体介导的攻击是特别敏感的。除了复发性奈瑟菌属感染,有原因不明的复发性化脓性细菌感染的患者也应该对其他更普遍的免疫缺陷进行检验,包括免疫球蛋白或吞噬细胞缺陷检测,它们比补体缺陷更普遍。
一些特殊临床表现(警示迹象)高度提示补体缺乏症。[10] 其中包括:
5 岁以上个体患流行性脑脊髓膜炎
其他复发性细菌感染,尤其是肺炎球菌
无荨麻疹的血管性水肿
自身免疫性表现
肾脏和眼系统的炎症性疾病
生殖器外淋球菌性感染。
通常为常染色体隐性遗传;备解素缺乏症(X 连锁遗传)、因子 B、C1-抑制因子和 MCP/CD46 缺乏症除外,它们为常染色体显性遗传。除年龄相关性黄斑变性和某些肾病(非典型溶血尿毒综合征、C3 肾小球疾病)外,杂合子携带者通常无临床症状。遗传性补体缺乏症可通过获取准确的病史并对整个家庭进行扩展实验室检查来进行确认。
C1-抑制因子缺乏和血管性水肿
无荨麻疹的血管性水肿占血管性水肿临床病例的约 2%。由于 C1-抑制因子缺乏,大约 50,000 人中有 1-10 人患有遗传性血管性水肿(HAE;也称为 Quincke 水肿)。HAE 表现为嘴唇、眼睑和胃肠道发送急性、阵发性肿胀(腹痛),如果肿胀发生在喉部和咽部则可能特别危险。肠梗阻、尿潴留和头痛可由其他器官水肿引起。压力、创伤相关感染以及由于妊娠、避孕、月经或激素替代治疗引起的激素变化都可引发血管性水肿。发作持续 1 周,如不进行治疗,25%-40% 的病例可导致窒息。
有两种常染色体显性形式的 HAE 与 C1-抑制因子缺乏 (C1-INH-HAE) 相关。在 80% 的病例中发现 C1-抑制因子产生不足 (HAE-1),而合成功能失调的 C1-抑制因子 (HAE-2) 占剩余的 20%。[15] 这两种形式典型的特点表明经典途径失调的继发结果是 C4 降低。经 C1-抑制因子的功能检测,两种形式的补体系统都是异常的,但是 HAE-2 中 C1-抑制因子的生物化学水平是正常的。[16] 缓激肽是血管渗漏导致血管性水肿的关键介导因素。获得性血管性水肿 (C1-INH-AAE) 是在 C1-抑制因子的过度分解代谢或自身抗体阻断后发生的。[17]
可通过血浆中降低的 C4 和降低的 C1-抑制因子水平来确定 HAE-1。在 HAE-2 中,C1-抑制因子的血浆水平通常是正常的,但其功能会显著降低。[16] 更罕见的获得性自身免疫性血管性水肿(C1q 活性降低)也会导致在 C1-抑制因子水平正常情况下,其功能显著降低。[16]
许多患者的临床表现与 HAE 相似,但未表现出 C1-抑制剂缺陷,且 C4 水平正常 (nlC1-INH-HAE) 。在这些患者中已观察到 FXII 基因 (FXII-HAE) 中的功能获得性突变、血浆纤维蛋白溶酶原缺乏 (PLG-HAE),以及不太常见的血管生成素-1 缺乏。[18]
C1-抑制因子和 C1q 也可因大量免疫复合物性疾病,如荨麻疹性血管炎,而被消耗。
其中一些病例有明确的家族病史。然而,尽管一些家属成员具有与其亲属相同的基因缺陷,也可能没有任何临床表现。
经典途径组分缺乏和 SLE
预先存在的补体缺乏,或包括 C1q、C4、C2、C3 在内的经典途径 (CP) 组分功能障碍,或补体受体(包括 C3b 受体)功能障碍,可诱发 SLE。最普遍和最严重的 SLE 与 C1、C2 或 C4 缺乏有关。[19] 缺乏其中一种蛋白质的人群超过四分之三都患有 SLE - 在缺乏 C1q 的病例中几率超过 90%。[19] 大约 10% 的 C2 缺乏患者患有 SLE。[19] 针对 C1q 的自身抗体(在 SLE 患者中普遍存在)可能可中和 C1q 功能且被认为具有预后价值。狼疮性肾炎是 SLE 的常见并发症。补体介导的免疫复合物消除的减少和细胞凋亡是该疾病的特征,并且在 C1q 基因缺乏的患者中最明显。[20]
监测 CP 溶血功能、C3 和 C4 的生化水平、活化产物和抗 C1q 自身抗体对一些 SLE 患者有所帮助(例如,当 SLE 引起肾小球肾炎时,临床改善通常伴随着 C3 和 C4 水平恢复正常)。
播散型奈瑟菌属感染中的补体缺乏症
C3、补体途径的末端组分 (C5-C9) 和旁路途径 (AP) 蛋白备解素的缺乏与侵入性脑膜炎球菌感染的发病率增加密切相关,尤其是罕见的血清型(W、X、Y、Z)。[21] 这表明细胞溶解补体活性对奈瑟球菌宿主防御的重要性。为识别这些缺乏,必须通过使用溶血分析(CH50、AH50)或功能性酶联免疫吸附测定 (ELISA) 对经典和旁路途径(也包括凝集素途径)的功能活性进行全面筛查分析。伴有复发性脑膜炎球菌感染的正常的 AH50 和 CH50 水平可能提示备解素缺乏。
强烈建议患者使用四价结合疫苗对脑膜炎奈瑟菌进行免疫接种。应该注意的是,并非所有与疾病相关的血清型都包括在这类疫苗中。[22]
肾病患者补体失调
补体与肾小球肾炎和终末期肾病的发病机制相关;[23][24] 肾脏特别容易受到补体介导的损伤。几种罕见类型的肾小球肾炎,包括致密沉积物病 (DDD) 和 C3 肾小球肾炎 (C3GN),都被归类为涵盖性术语 C3 肾小球疾病 (C3G)。这组肾脏疾病是由补体活化的异常控制引起的,其中补体组分 C3 在肾小球中沉积,导致可变的细胞炎症。[25] C3 肾炎因子是 C3 稳定抗体,可见于 40%-60% 的 C3GN 病例和 80%-90% 的 DDD 病例。[25] C3G 的患者,尤其是 DDD 的患者,通常 CH50、AH50 和 C3 水平低。
非典型溶血尿毒综合征的特征为微血管病溶血性贫血、血小板减少症和急性肾衰竭。[26] 大多数病例与导致补体活化失调的突变或自身抗体有关。[26] 由产志贺毒素大肠埃希菌引起的腹泻阳性溶血尿毒综合征 (HUS) 中也会偶尔发现补体基因的突变。[26] 有非典型性溶血性尿毒综合征的患者可能有非典型性溶血性尿毒综合征的阳性家族病史。
诊断包括总溶血活性 (CH50、AH50)、C3、C3 活化产物(C3a 或 C3d)和 sC5b-9 的分析,以及 C3、CFB、CFH、CHFR1-5、CFI、MCP/CD46 和 THBD/CD141 的分子遗传学分析。C3 水平减低且 C4 水平正常可能已经指向各种形式的肾病(如 aHUS、DDD)中存在旁路途径缺陷,如因子 H 和 I 缺乏或自身抗体(如 C3 NeF、抗因子 H)缺乏。
年龄相关性黄斑变性
补体级联反应的旁路途径失调似乎在年龄相关性黄斑变性 (AMD) 的发病机制中具有关键作用。已在患有 AMD 的人的视网膜沉积物中发现各种补体蛋白、它们的活化产物和调节因子。编码补体系统蛋白(尤其是调节因子 H,以及 C3、C2、因子 B 和因子 I)的基因中的病理诱导多态性与 AMD 相关。[27]
阵发性睡眠性血红蛋白尿
阵发性睡眠性血红蛋白尿症 (PNH) 是一种罕见的后天获得性疾病,一般发生于成人(女性多于男性),表现为溶血、血栓形成以及全血细胞减少三联征。它是由 PIGA 基因突变引起的红细胞克隆性疾病。[28] 由于 PIGA 功能缺陷,受影响的红细胞缺乏所有糖基磷脂酰肌醇连接的膜蛋白,包括 CD55 和 CD59。CD59 和 CD55 的缺失使得 PNH 红细胞容易受到自体 C 介导的裂解的影响,从而导致溶血性贫血。
孤立性 CD55 缺乏很罕见,但已见于患有严重早发性蛋白丢失性肠病的患者。除了溶血外,严重的吉兰-巴雷综合征样神经系统症状是孤立性 CD59 缺乏的标志性症状。
冷球蛋白血症
血液系统恶性肿瘤可引起冷球蛋白血症。它会导致经典途径活化,补体 C4 减少。[29] C3 和 C4 水平正常不能排除冷球蛋白血症。如果临床怀疑疾病与补体 C4 减少有关时,应检测血清冷凝蛋白水平(例如,当患者出现血管炎性皮疹,较冷的四肢尤为严重;关节炎;或急性肾衰竭)。确定性诊断检测包括获得新鲜凝血,收集到预热 (37°C [98.6°F]) 的保温瓶中;然后快速分离血清以降低冷凝球蛋白留在血凝块中的风险。
部分性脂肪代谢障碍
部分脂肪代谢障碍通常与自身抗体 C3 肾炎因子有关。C3 转化酶稳定会导致脂肪细胞的补体持续活化和裂解。[30]
应对有以下情况的患者进行可能的补体缺乏症检测:
风湿性免疫疾病(例如,SLE和其他自身免疫结缔组织疾病)
无荨麻疹的血管性水肿
肾脏疾病
传染病(例如,非典型性或复发的细菌感染,如罕见脑膜炎球菌血清型病例以及复发性脑膜炎球菌脓毒症)
部分性脂肪代谢障碍
冷球蛋白血症
补体缺乏症家族病史。
临床表现可提示患者的潜在补体缺乏,以及应该施行哪种血液检测。
潜在补体缺乏症的分析应从每个活化途径的功能筛查开始,然后从功能、蛋白质和分子水平方面鉴别缺陷。[31] 鉴于补体蛋白在广泛生物系统中的重要性,需要进行有效、标准化和可执行的检测。实验室之间在补体检测方面的质量差异很大,只有少数检测可商用。但是,一些专业实验室可提供比传统的 C3 和 C4 参数更先进的综合诊断。European diagnostic complement labs
必须将血液样本分离为血清和乙二胺四乙酸 (EDTA) 血浆。血清足以分析总功能、补体蛋白和调节因子以及自身抗体。EDTA 对于活化产物的定量是绝对必需的。最终浓度≥10 mM 的 EDTA 用作标准抗凝血剂,因为其 Mg2+ 和 Ca2+ 络合特性阻断了补体系统的体外活化。[10] 肝素化和含枸橼酸盐的血液作用不大。如果未能在几小时内对血清/血浆样本进行分析,则应将其深度冷冻并用干冰快递送至专业实验室。由于存在体外活化风险,它们不应进行反复冻融循环。
整体检测(例如溶血性 CH50 和 AH50 或基于脂质体的 CP 裂解检测)可提供有关整个补体级联反应如何起作用的信息。这些检测方法需要人工测量试管中的靶细胞裂解情况。检测取决于引起靶细胞裂解的膜攻击复合物 (C5b-C9) 的完全组装。活性缺失或显著减少表明患原发性补体缺乏症。但是,这一发现也可能是由于补体消耗增加而导致的继发性缺乏症。[10] C3 水平降低且 C4 水平正常指示旁路途径 (AP) 调节因子缺陷,例如因子 H 和 I 缺乏,或 C3 肾病因子诱导的 AP 失调,也见于各种形式的肾病(例如,非典型溶血尿毒综合征 [aHUS]、C3 肾小球病 [C3G]/致密沉积物病 [DDD])。
为快速分析缺乏,已开发了一种酶联免疫吸附测定 (ELISA),它可以同时检测所有三种活化途径。[32] 随后需要对组分进行单独分析以确定补体级联反应(过度)活化发生的部分。补体活化产物(如 C3a、C3d 或 sC5b-9)的分析对区分是否由过度活化引起的原发性和继发性补体缺乏症而言是必需的,并且有助于确定补体靶向治疗(例如,艾库组单抗治疗)的疗效。[33]
使用此内容应接受我们的免责声明。
