确切的病因尚不清楚,但似乎与肝脏细胞内有机阴离子包括胆红素二葡糖苷酸在内的储存容量缺陷有关。[8]Zimniak P. Dubin-Johnson and Rotor syndromes: molecular basis and pathogenesis. Semin Liver Dis. 1993;13:248-260.http://www.ncbi.nlm.nih.gov/pubmed/8235715?tool=bestpractice.com[9]Wolpert E, Pascasio FM, Wolkoff AW. Abnormal sulfobromophthalein metabolism in Rotor's syndrome and obligate heterozygotes. N Engl J Med. 1977;296:1099-1101.http://www.ncbi.nlm.nih.gov/pubmed/850521?tool=bestpractice.com
对 8 个罗托综合征 (RS) 家庭的分析发现,该病预测会导致完全且同步的有机阴离子转运多肽 OATP1B1 和 OATP1B3 缺陷的突变有关。这些重要的解毒限制蛋白质将介导窦状肝细胞膜中无数药物及药物复合体的吸收和清除。该研究表明在 RS 病例,某些药物可引起威胁生命的毒性风险。[10]van de Steeg E, Stránecký V, Hartmannová H, et al. Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. J Clin Invest. 2012;122:519-528.http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3266790/?tool=pubmedhttp://www.ncbi.nlm.nih.gov/pubmed/22232210?tool=bestpractice.com
已识别出四种胆红素代谢遗传缺陷:Gilbert 综合征和 Crigler-Najjar 综合征(I 型和 II 型)与非结合型高胆红素血症相关,而 Dubin-Johnson 综合征 (DJS) 和 RS 与结合型高胆红素血症相关。胆红素是血红素分解代谢的副产物。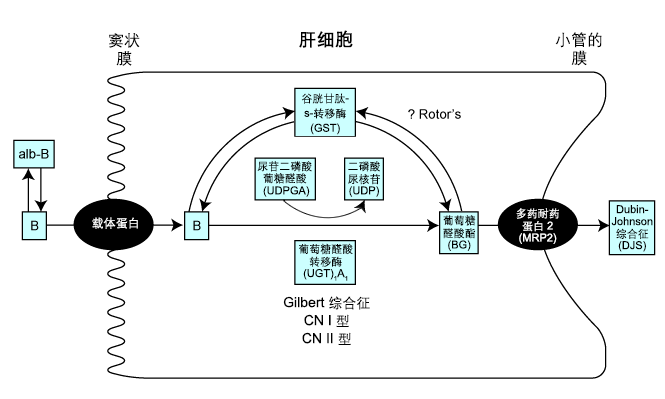 [Figure caption and citation for the preceding image starts]: 肝细胞内胆红素 (B) 代谢的示意图由 Dr Coelho 提供 [Citation ends]. 胆红素 (B) 的正常处置涉及其通过白蛋白载体 (alb-B) 向肝脏的转运。在肝细胞中,它同糖分子、 葡萄糖醛酸 (G) 结合成胆红素葡萄糖醛酸酯 (BG),为一种可通过粪便轻易排出的水溶性化合物。供体则是尿苷二磷酸葡糖醛酸 (UDPGA),而这一反应由尿苷二磷酸葡萄糖醛酸转移酶(UGT1A1-特异性异构体)催化。这种酶的活性降低与 Gilbert 综合征和 Crigler-Najjar 综合征(I 型和 II 型)有关。谷胱甘肽-s-转移酶 (GST) 为某些有机分子充当细胞内载体蛋白。[11]Tipping E, Ketterer B. The role of intracellular proteins in the transport and metabolism of lipophilic compounds. In: Blaver G, Sund H, eds. Transport by proteins. Berlin: Walter de Gruyter & Co.; 1978:369.有人认为RS 患者可能存在肝 GST 缺乏。[12]Adachi Y, Yamamoto T. Partial defect in hepatic glutathione S-transferase activity in a case of Rotor's syndrome. Gastroenterol Jpn. 1987;22:34-38.http://www.ncbi.nlm.nih.gov/pubmed/2952540?tool=bestpractice.com这种缺乏将可能导致胞液内胆红素吸收受损。此外,胆红素结合物在等待从肝细胞中排泄到小管腔时将同 GST 结合,因此细胞内储存缺乏会导致胆红素结合物渗漏回到循环中。[13]Wolkoff AW, Ketley JN, Waggoner JG, et al. Hepatic accumulation and intracellular binding of conjugated bilirubin. J Clin Invest. 1978;61:142-149.http://www.jci.org/articles/view/108912http://www.ncbi.nlm.nih.gov/pubmed/618909?tool=bestpractice.com小管多药耐药蛋白 2 (MRP2) 似乎对胆红素的小管分泌很重要。MRP2 的缺陷将导致 DJS。
[Figure caption and citation for the preceding image starts]: 肝细胞内胆红素 (B) 代谢的示意图由 Dr Coelho 提供 [Citation ends]. 胆红素 (B) 的正常处置涉及其通过白蛋白载体 (alb-B) 向肝脏的转运。在肝细胞中,它同糖分子、 葡萄糖醛酸 (G) 结合成胆红素葡萄糖醛酸酯 (BG),为一种可通过粪便轻易排出的水溶性化合物。供体则是尿苷二磷酸葡糖醛酸 (UDPGA),而这一反应由尿苷二磷酸葡萄糖醛酸转移酶(UGT1A1-特异性异构体)催化。这种酶的活性降低与 Gilbert 综合征和 Crigler-Najjar 综合征(I 型和 II 型)有关。谷胱甘肽-s-转移酶 (GST) 为某些有机分子充当细胞内载体蛋白。[11]Tipping E, Ketterer B. The role of intracellular proteins in the transport and metabolism of lipophilic compounds. In: Blaver G, Sund H, eds. Transport by proteins. Berlin: Walter de Gruyter & Co.; 1978:369.有人认为RS 患者可能存在肝 GST 缺乏。[12]Adachi Y, Yamamoto T. Partial defect in hepatic glutathione S-transferase activity in a case of Rotor's syndrome. Gastroenterol Jpn. 1987;22:34-38.http://www.ncbi.nlm.nih.gov/pubmed/2952540?tool=bestpractice.com这种缺乏将可能导致胞液内胆红素吸收受损。此外,胆红素结合物在等待从肝细胞中排泄到小管腔时将同 GST 结合,因此细胞内储存缺乏会导致胆红素结合物渗漏回到循环中。[13]Wolkoff AW, Ketley JN, Waggoner JG, et al. Hepatic accumulation and intracellular binding of conjugated bilirubin. J Clin Invest. 1978;61:142-149.http://www.jci.org/articles/view/108912http://www.ncbi.nlm.nih.gov/pubmed/618909?tool=bestpractice.com小管多药耐药蛋白 2 (MRP2) 似乎对胆红素的小管分泌很重要。MRP2 的缺陷将导致 DJS。
RS为常染色体隐性遗传。在一项研究中,RS 与其他遗传性疾病(葡萄糖-6-磷酸脱氢酶 [G6PD] 缺乏症以及杂合性 β 地中海贫血)共存,提示在共同遗传的基因之间存在可能的相互作用。[7]Fretzayas A, Koukoutsakis P, Moustaki M, et al. Coinheritance of Rotor syndrome, G-6-PD deficiency, and heterozygous beta thalassemia: a possible genetic interaction. J Pediatr Gastroenterol Nutr. 2001;33:211-213.http://journals.lww.com/jpgn/pages/articleviewer.aspx?year=2001&issue=08000&article=00023&type=fulltexthttp://www.ncbi.nlm.nih.gov/pubmed/11568527?tool=bestpractice.com
