病因学
在原发性肌张力障碍中,尚未发现结构性病变和相关毒素可导致肌张力障碍发生,尽管在某些病例中,可以确定由基因突变引发肌张力障碍,特别是一些早发型或家族性病例。 但是,大多数原发性肌张力障碍都是特发性的。[2][8][9][10][11][12][13] 由于外显率较低,遗传性肌张力障碍并非都有家族史。 若干遗传因素已被确定与肌张力障碍相关。 这些遗传因素包括常染色体显性(外显率不完全)、常染色体隐性、X-连锁和线粒体遗传病因。[13]
早发型肌张力障碍患者(通常 26 岁以前发病)中常常可发现携带编码 TorsinA 蛋白的基因 DYTI 发生突变,其典型表现为单个肢体局灶性起病,且逐渐发展为全身性。[14]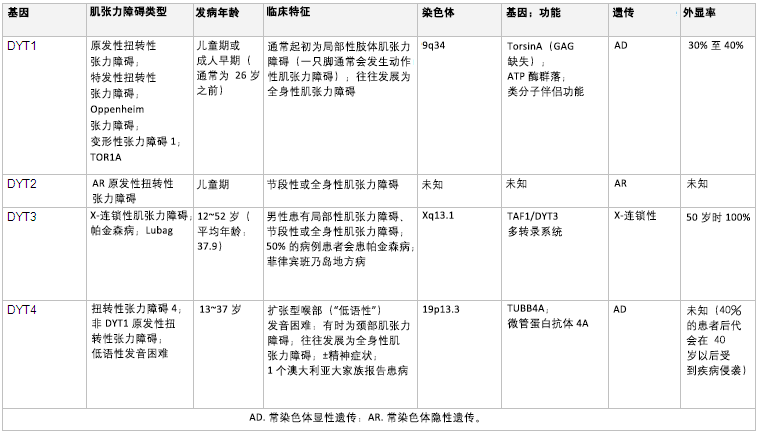 [Figure caption and citation for the preceding image starts]: 遗传学和肌张力障碍摘自 N Engl J Med. 2006 Aug 24;355(8):818-29; 经授权使用。更多信息请参阅”运动障碍 (Mov Disord)“。2011 May;26(6):1106-26 [Citation ends].
[Figure caption and citation for the preceding image starts]: 遗传学和肌张力障碍摘自 N Engl J Med. 2006 Aug 24;355(8):818-29; 经授权使用。更多信息请参阅”运动障碍 (Mov Disord)“。2011 May;26(6):1106-26 [Citation ends].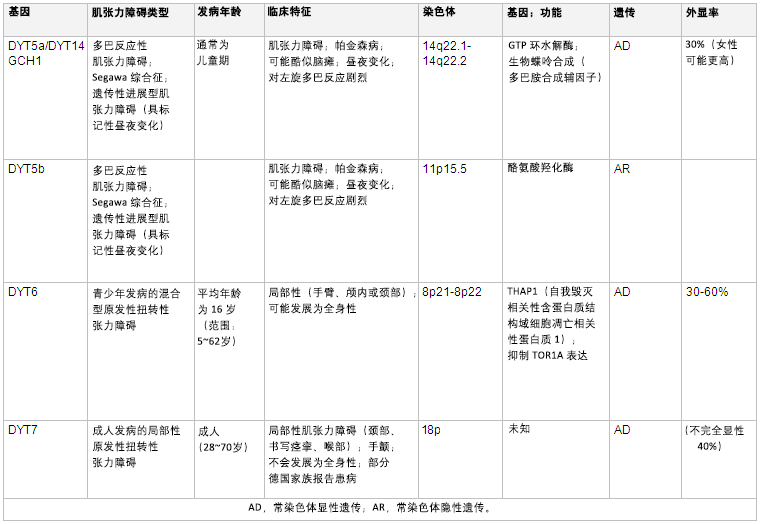 [Figure caption and citation for the preceding image starts]: 遗传学和肌张力障碍摘自 N Engl J Med. 2006 Aug 24;355(8):818-29; 经授权使用。更多信息请参阅”运动障碍 (Mov Disord)“。2011 May;26(6):1106-26 [Citation ends].
[Figure caption and citation for the preceding image starts]: 遗传学和肌张力障碍摘自 N Engl J Med. 2006 Aug 24;355(8):818-29; 经授权使用。更多信息请参阅”运动障碍 (Mov Disord)“。2011 May;26(6):1106-26 [Citation ends]. [Figure caption and citation for the preceding image starts]: 遗传学和肌张力障碍摘自 N Engl J Med. 2006 Aug 24;355(8):818-29; 经授权使用。更多信息请参阅“运动障碍 (Mov Disord)“。2011 May;26(6):1106-26 和 Neuropath Applied Neurobiol。2012 Oct;38(6):520-34 [Citation ends].
[Figure caption and citation for the preceding image starts]: 遗传学和肌张力障碍摘自 N Engl J Med. 2006 Aug 24;355(8):818-29; 经授权使用。更多信息请参阅“运动障碍 (Mov Disord)“。2011 May;26(6):1106-26 和 Neuropath Applied Neurobiol。2012 Oct;38(6):520-34 [Citation ends].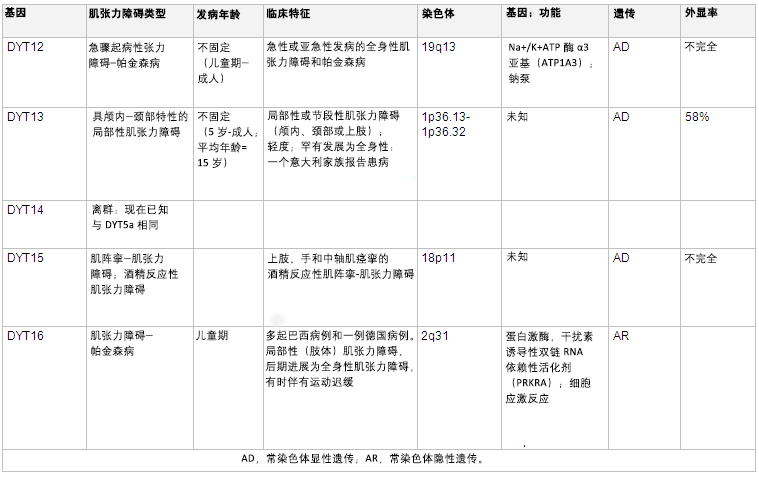 [Figure caption and citation for the preceding image starts]: 遗传学和肌张力障碍摘自 N Engl J Med. 2006 Aug 24;355(8):818-29; 经授权使用。更多信息请参阅”运动障碍 (Mov Disord)“。2011 May;26(6):1106-26 [Citation ends].
[Figure caption and citation for the preceding image starts]: 遗传学和肌张力障碍摘自 N Engl J Med. 2006 Aug 24;355(8):818-29; 经授权使用。更多信息请参阅”运动障碍 (Mov Disord)“。2011 May;26(6):1106-26 [Citation ends]. [Figure caption and citation for the preceding image starts]: 遗传学和肌张力障碍摘自 N Engl J Med. 2006 Aug 24;355(8):818-29; 经授权使用。更多信息请参阅 Mov Disord. 2011 May;26(6):1106-26 和 Mov Disord. 2013 Jun 15;28(7):899-905 [Citation ends].
[Figure caption and citation for the preceding image starts]: 遗传学和肌张力障碍摘自 N Engl J Med. 2006 Aug 24;355(8):818-29; 经授权使用。更多信息请参阅 Mov Disord. 2011 May;26(6):1106-26 和 Mov Disord. 2013 Jun 15;28(7):899-905 [Citation ends].
继发性肌张力障碍可能有以下病因:
基底神经节受累的结构性损伤(中风、肿瘤、感染);此外还报告有丘脑、脑干、脑皮层或小脑损伤病例。
伴随其他神经系统疾病(如帕金森病、肝豆状核变性和脑瘫)。 围产期脑损伤同时伴有早期生长发育里程的异常更提示继发性肌张力障碍(脑瘫),而非原发性的。
急性或慢性服用典型和非典型抗精神病药、多巴胺受体阻断剂和止吐剂(如甲氧氯普胺和丙氯拉嗪),与药物诱导性肌张力障碍具有强烈相关性。 药物诱发的肌张力障碍可能是急性肌张力障碍反应或迟发性肌张力障碍。
创伤;如果病因是头部创伤,创伤后肌张力障碍通常伴有其他神经系统症状,包括震颤、无力和痉挛。 伴有反射性交感神经营养障碍症状的相对速发型固定肌张力障碍被认为是另一种形式的创伤后肌张力障碍,通常是局部性肢体肌张力障碍。[15] 这种形式的肌张力障碍通常对原发性肌张力障碍所用的疗法的反应不佳。 许多病例报道局部身体创伤性损伤后的几天或几周后,在损伤部位出现类似局部肌张力障碍症状的表现,尽管创伤和肌张力障碍之间的确切病因关系尚存在争论。[16][17][18]
运动;有说服力的事例证明,需要频繁动用肌张力障碍性部位参与来完成某个特定动作的人比不经常使用该部位的人更容易出现该部位的局部性肌张力障碍(如音乐家肌张力障碍)。
肌张力障碍也是更复杂的泛发型神经退行性障碍的一种临床特征,有时被称为“遗传性退行性”肌张力障碍。[2][8][9][10][11] 它们也可能是阵发性或精神性的。
早发型肌张力障碍往往被认为具有与晚发型肌张力障碍完全不同的病因。 早发型全身性肌张力障碍与 DYT1 突变或多巴反应性肌张力障碍具有相关性,在 26 岁以下患者中发病的可能性更高,通常会在儿童早期发病。[19]
若干诊断算法已被提出,用于确保正确评估童年及青春期肌张力障碍。[20][21] 第一步是排除与童年及青春期肌张力障碍相似的疾病;第二步是排除药物诱发的肌张力障碍;第三步是评估出现获得性肌张力障碍的可能性;最后一步是在考虑新一代测序技术之前,建议先进行生化和代谢检查。
病理生理学
病理生理学非常复杂,在不同条件下有不同的因素可以导致肌张力障碍的发生。 肌张力障碍的病生理机制仍不明确。 多巴反应性肌张力障碍中的多巴胺缺乏和左旋多巴有效提示多巴胺可能参与某些类型肌张力障碍的发生和发展。 肌张力障碍与帕金森病所具有的密切相关性,以及多巴胺阻断抗精神病药诱发肌张力障碍的特性进一步证实了多巴胺在肌张力障碍发病中所起的作用。[2][9]
正电子发射体层扫描(PET)和苍白球电位记录已经检测到了基底神经节神经元电活动的异常。[22] 正电子发射断层显像 (PET) 和荧光-磁共振成像通常会显示运动前区皮层和豆状核内静息状态下葡萄糖代谢增加,以及运动前区皮层和初级运动皮层内可能的异常电活动。电生理学检查显示中枢神经系统抑制活动受损。受损的周围抑制可能导致神经活动扩散至邻近激活的神经回路区域,从而有可能导致运动信号不当溢流至邻近肌肉。局灶性肌张力障碍受累身体部位的感觉运动表现会被大脑皮层放大。但是,目前仍不清楚这些变化是原发性的还是继发性的。[9][12]
分类
肌张力障碍分类:临床特点和病因[1]
由于新出现的病因和旧命名法存在的问题导致专家共识委员会制订和发布更新的肌张力障碍分类。 肌张力障碍现在根据两个主轴进行分类:临床表现和生物学病因。
轴线 I. 临床特点
肌张力障碍综合征的根据年龄的二分法(儿童发病型或成人发病型)现已被五分法取代,根据发病年龄不同,划分为五个不同的年龄段。 根据受累体内分布划分的类型包括局部性、节段性、多灶性、全身性和偏身性肌张力障碍。
新分类还区别了各种时间形式的肌张力障碍。 病程可以是稳定性或进展性的,疾病变异性包括持续性、动作特异性、昼行性和阵发性模型。
伴随特征非常重要。 肌张力障碍可以孤立出现,或者伴有其他神经系统特征(联合性肌张力障碍)。 孤立性肌张力障碍包括以前被称为“原发性”肌张力障碍的肌张力障碍,但是震颤除外,且肌张力障碍是唯一的运动异常。 联合性肌张力障碍是伴有其他运动障碍疾病(如帕金森病或肌阵挛)的肌张力障碍。
在 2013 年发布的分类共识中,“孤立性”是指现象,而非病因。 在“联合性”肌张力障碍中,肌张力障碍不一定是主要的运动障碍。
轴线 II. 病因
包括遗传性和获得性疾病,其中有退行性脑病、获得性结构性脑损伤或无可识别的大脑异常的疾病。遗传性疾病包括常染色体显性、常染色体隐性、X-连锁和线粒体病。当前基因 (DYT) 分类保留了亚型,但其不作为分类系统。它包括已证实的和未经证实的基因位点异常,往往与病因和临床表型具有不确定的相关性。DYT1 至 DYT25 基因位点包括一组混杂的肌张力障碍综合征,其中的部分疾病极为罕见,可能仅限于单个家族。引发肌张力障碍的获得性疾病包括创伤性、感染性、药物诱发性、中毒性、血管性、肿瘤性和精神性病因。散发性或家族性特发性成人发病型局灶性或节段性肌张力障碍亦被纳入Axia II 中。
 [Figure caption and citation for the preceding image starts]: 肌张力障碍的分类共识经作者 Albanese 等人允许后再版。Mov Disord.2013 Jun 15;28(7):863-73 [Citation ends].
[Figure caption and citation for the preceding image starts]: 肌张力障碍的分类共识经作者 Albanese 等人允许后再版。Mov Disord.2013 Jun 15;28(7):863-73 [Citation ends].
使用此内容应接受我们的免责声明。
