在任何年龄,HS 通常均可被轻易地诊断出来。在儿童中,最常被诊断为轻度疾病,在这种情况下可能存在黄疸(严重程度随时间推移而存在差异)和脾肿大。常规实验室检查通常通过检测以下方面来证实诊断:
具有非典型特征的人群可能需要接受验证性的实验室检查。[14]King MJ, Garçon L, Hoyer JD, et al; International Council for Standardization in Haematology (ICSH). ICSH guidelines for the laboratory diagnosis of nonimmune hereditary red cell membrane disorders. Int J Lab Hematol. 2015;37:304-325.http://onlinelibrary.wiley.com/doi/10.1111/ijlh.12335/fullhttp://www.ncbi.nlm.nih.gov/pubmed/25790109?tool=bestpractice.com 罕见情况下需要额外的专科检查。当怀疑诊断为 HS 时,应将患者转诊至血液科专科医师处接受确诊和常规监测。
 [Figure caption and citation for the preceding image starts]: HS 诊断流程由 BMJ Evidence Centre 使用来自作者的信息编制 [Citation ends].
[Figure caption and citation for the preceding image starts]: HS 诊断流程由 BMJ Evidence Centre 使用来自作者的信息编制 [Citation ends].
病史
HS 是北欧裔白种人溶血的最常见原因。HS 可见于多数种族人群,但在黑人中较少见。[15]Bolton-Maggs PH, King MJ. Hereditary spherocytosis, hereditary elliptocytosis and related disorders. In: Young NS, Gershon SL, High KA, eds. Clinical hematology. Philadelphia, PA: Mosby Elsevier; 2006:293-307. 多数 HS 患者会有贫血、黄疸、脾切除术或已知 HS 的家族史。
症状可在任何年龄出现。受累较严重的人往往会在生命较早期出现症状。临床严重程度的范围包括从无症状到严重贫血和黄疸引起的乏力。当没有症状时,经常在出于另一项原因进行血液检查时,偶然发现该疾病。
儿童或成人可能无症状,直到他们感染细小病毒 B19,发生由此导致的再生障碍危象。[16]Young NS. Parvovirus infection and its treatment. Clin Exp Immunol. 1996;104(suppl 1):26-30.http://www.ncbi.nlm.nih.gov/pubmed/8625539?tool=bestpractice.com 该病毒直接攻击骨髓中的红系前体细胞,导致红细胞再生障碍持续约 10 天。红细胞寿命缩短(例如见于 HS 和其他溶血性疾病患者)的患者在缺乏红细胞生成期间出现快速进展性贫血,并且通常表现为急性发作的明显面色苍白、嗜睡和发热。血红蛋白 (Hb) 值通常介于 3-6 g/dL。网织红细胞计数通常 <1%。在恢复阶段,网织红细胞计数增加,并且在从再生障碍性危象恢复后,通常会导致终身免疫。这种感染可能促使发现家庭中迄今未确诊的 HS 患者。
症状和体征可能随着时间的推移而变化。与再生障碍性发作相比,高溶血性危象更常见,但严重程度较轻,其特征在于加速常见的溶血过程,从而导致症状恶化。这些发作通常伴有非特异性病毒感染,在这些感染情况下,网状内皮系统出现增生,且脾脏进一步增大。可能会反复发作。 [Figure caption and citation for the preceding image starts]: HS 的表现(按年龄)由 BMJ Evidence Centre 使用来自作者的信息编制 [Citation ends].
[Figure caption and citation for the preceding image starts]: HS 的表现(按年龄)由 BMJ Evidence Centre 使用来自作者的信息编制 [Citation ends].
体格检查
许多 HS 患者有轻度黄疸。当 HS 出现在新生儿期时,黄疸是一种常见的特征,其严重程度不一定与 HS 的未来严重程度相关。在某些情况下,黄疸会严重到需要进行换血疗法。[5]Bolton-Maggs PH, Langer JC, Iolascon A, et al; General Haematology Task Force of the British Committee for Standards in Haematology. Guidelines for the diagnosis and management of hereditary spherocytosis - 2011 update. Br J Haematol. 2012;156:37-49.http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2141.2011.08921.x/fullhttp://www.ncbi.nlm.nih.gov/pubmed/22055020?tool=bestpractice.com 极少数情况下,HS 可能表现为由严重贫血引起的胎儿水肿或死胎(当婴儿遗传来自双亲的膜蛋白缺陷时)。[9]Gallagher PG, Weed SA, Tse WT, et al. Recurrent fatal hydrops fetalis associated with a nucleotide substitution in the erythrocyte beta-spectrin gene. J Clin Invest. 1995;95:1174-1182.http://www.jci.org/articles/view/117766/pdfhttp://www.ncbi.nlm.nih.gov/pubmed/7883966?tool=bestpractice.com[10]Ribeiro ML, Alloisio N, Almeida H, et al. Severe hereditary spherocytosis and distal renal tubular acidosis associated with the total absence of band 3. Blood. 2000;96:1602-1604.http://www.bloodjournal.org/content/96/4/1602.fullhttp://www.ncbi.nlm.nih.gov/pubmed/10942416?tool=bestpractice.com[11]Whitfield CF, Follweiler JB, Lopresti-Morrow L, et al. Deficiency of alpha-spectrin synthesis in burst-forming units-erythroid in lethal hereditary spherocytosis. Blood. 1991;78:3043-3051.http://www.bloodjournal.org/content/bloodjournal/78/11/3043.full.pdfhttp://www.ncbi.nlm.nih.gov/pubmed/1954389?tool=bestpractice.com
对于有意外或不明原因脾肿大的任何年龄患者,都应考虑 HS 的诊断。脾肿大(轻度至中度肿大)在任何严重程度的 HS 患者中均非常常见,但并非这种疾病所特有的。[5]Bolton-Maggs PH, Langer JC, Iolascon A, et al; General Haematology Task Force of the British Committee for Standards in Haematology. Guidelines for the diagnosis and management of hereditary spherocytosis - 2011 update. Br J Haematol. 2012;156:37-49.http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2141.2011.08921.x/fullhttp://www.ncbi.nlm.nih.gov/pubmed/22055020?tool=bestpractice.com 它由异常、无弹性的红细胞在脾脏内被诱捕和破坏引起。脾肿大通常没有任何症状或临床后果。然而,在高溶血性危象期间,脾脏可能罕见地急剧显著肿大,从而引起左上腹疼痛和早饱的症状。脾脏通常会在发作间期缩小到之前的大小。
患有 HS 的个体可能有也可能没有贫血的体征。根据病情的严重程度,贫血的体征存在差异:从没有明显的体征到严重苍白。 [Figure caption and citation for the preceding image starts]: HS 患儿眼部巩膜黄染由英国曼彻斯特大学 (University of Manchester) Paula Bolton-Maggs 提供;经获准使用 [Citation ends].
[Figure caption and citation for the preceding image starts]: HS 患儿眼部巩膜黄染由英国曼彻斯特大学 (University of Manchester) Paula Bolton-Maggs 提供;经获准使用 [Citation ends].
初始检查
许多病例是在因其他原因接受血液检查时被偶然发现的。对于任何有黄疸或脾肿大或者有一级亲属 HS 家族史的患者,也应考虑该诊断。[17]Christensen RD, Yaish HM, Gallagher PG. A pediatrician's practical guide to diagnosing and treating hereditary spherocytosis in neonates. Pediatrics. 2015;135:1107-1114.http://www.ncbi.nlm.nih.gov/pubmed/26009624?tool=bestpractice.com
无论任何年龄,在血红蛋白结果意外低的情况下,应考虑该诊断。大多数情况下,血细胞计数和涂片具有诊断意义。患者的血涂片显示血红蛋白正常或减少、球形红细胞(异常形态的红细胞),并且网织红细胞增多。除了球形红细胞外,血涂片可能出现钳状细胞(蘑菇形细胞),这是由于带 3 蛋白突变所致。当骨髓输出的增加程度足以维持血红蛋白正常时,HS 患者可能不存在贫血,但网织红细胞将会增加(被称为代偿性溶血)。
仔细分析血涂片上的红细胞形态非常重要,这样做可以避免遗漏其他不太常见的疾病。[15]Bolton-Maggs PH, King MJ. Hereditary spherocytosis, hereditary elliptocytosis and related disorders. In: Young NS, Gershon SL, High KA, eds. Clinical hematology. Philadelphia, PA: Mosby Elsevier; 2006:293-307. 红细胞参数也可能与诊断一致,通常有平均红细胞体积 (mean corpuscular volume, MCV) 正常或减小,并且经常存在平均血红蛋白浓度 (mean corpuscular haemoglobin concentration, MCHC) 升高。球形红细胞不仅仅见于 HS 患者,必须通过适当的病史、临床情况和检查来排除其他诊断。[15]Bolton-Maggs PH, King MJ. Hereditary spherocytosis, hereditary elliptocytosis and related disorders. In: Young NS, Gershon SL, High KA, eds. Clinical hematology. Philadelphia, PA: Mosby Elsevier; 2006:293-307. 新生儿血涂片可能难以解读。如果诊断不明确,需要仔细回顾血涂片,并进行随访。在几个月后,更容易看到球形红细胞的典型血涂片表现。
球形细胞增多症最重要的替代诊断是自身免疫性溶血性贫血 (auto-immune haemolytic anaemia, AIHA)。在 AIHA 患者中,异常抗体会覆盖红细胞,通过直接抗人球蛋白试验 (DAT) 可以检测出来,而在 HS 患者中这一检测结果正常。在婴儿期,还必须排除由不规则母体 IgG 抗体所致溶血这一可能的原因。
鉴别 HS 与其他遗传性红细胞膜疾病很重要。其中许多症状在血涂片表现(例如,椭圆形红细胞增多症)中显而易见,但是在红细胞外观不典型的情况下,应该考虑其他诊断。排除具有膜运输缺陷的遗传性口腔细胞增多症尤其重要,因为脾切除术后具有特别高的静脉血栓栓塞风险。如果在临床上怀疑黄疸,则进行血清胆红素和肝脏转氨酶检测。先天性红细胞生成异常性贫血的病例也曾被误诊为 HS。 [Figure caption and citation for the preceding image starts]: HS (A) 患者的血涂片与正常血涂片 (B) 进行比较;箭头指示螯状细胞(蘑菇形细胞)由英国曼彻斯特大学 (University of Manchester) Paula Bolton-Maggs 提供;经获准使用 [Citation ends].
[Figure caption and citation for the preceding image starts]: HS (A) 患者的血涂片与正常血涂片 (B) 进行比较;箭头指示螯状细胞(蘑菇形细胞)由英国曼彻斯特大学 (University of Manchester) Paula Bolton-Maggs 提供;经获准使用 [Citation ends]. [Figure caption and citation for the preceding image starts]: HS 患者的血涂片;箭头指示球形红细胞由德克萨斯大学西南医学中心 (University of Texas Southwestern Medical Center) Shelley Crary 提供;获准后使用 [Citation ends].
[Figure caption and citation for the preceding image starts]: HS 患者的血涂片;箭头指示球形红细胞由德克萨斯大学西南医学中心 (University of Texas Southwestern Medical Center) Shelley Crary 提供;获准后使用 [Citation ends].
需考虑的确证试验
对于 HS 的典型病例,不需要任何确证性试验。当特征不典型时(例如,血涂片上的形态不是很典型或没有家族史),推荐使用伊红-5-马来酰亚胺 (EMA) 结合试验进行确认。[14]King MJ, Garçon L, Hoyer JD, et al; International Council for Standardization in Haematology (ICSH). ICSH guidelines for the laboratory diagnosis of nonimmune hereditary red cell membrane disorders. Int J Lab Hematol. 2015;37:304-325.http://onlinelibrary.wiley.com/doi/10.1111/ijlh.12335/fullhttp://www.ncbi.nlm.nih.gov/pubmed/25790109?tool=bestpractice.com EMA 结合试验基于通过流式细胞术测量的荧光强度。[18]King MJ, Behrens J, Rogers C, et al. Rapid flow cytometric test for the diagnosis of membrane cytoskeleton-associated haemolytic anaemia. Br J Haematol. 2000;111:924-933.http://www.ncbi.nlm.nih.gov/pubmed/11122157?tool=bestpractice.com[19]King MJ, Smythe JS, Mushens R. Eosin-5-maleimide binding to band 3 and Rh-related proteins forms the basis of a screening test for hereditary spherocytosis. Br J Haematol. 2004;124:106-113.http://www.ncbi.nlm.nih.gov/pubmed/14675415?tool=bestpractice.com[20]King MJ, Telfer P, MacKinnon H, et al. Using the eosin-5-maleimide binding test in the differential diagnosis of hereditary spherocytosis and hereditary pyropoikilocytosis. Cytometry B Clin Cytom. 2008;74:244-250.http://www.ncbi.nlm.nih.gov/pubmed/18454487?tool=bestpractice.com EMA 结合带 3 蛋白;与正常情况相比,在患 HS 的情况下,带 3 蛋白破坏会导致荧光减少。据报告,这对识别 HS 的敏感度为 92.7%,特异度为 99.1%,[18]King MJ, Behrens J, Rogers C, et al. Rapid flow cytometric test for the diagnosis of membrane cytoskeleton-associated haemolytic anaemia. Br J Haematol. 2000;111:924-933.http://www.ncbi.nlm.nih.gov/pubmed/11122157?tool=bestpractice.com 但在其他红细胞疾病(特别是先天性红细胞生成异常性贫血 II 型 [congenital dyserythropoietic anaemia type II, CDA-II])时也可能异常。
另一种验证性试验是酸化甘油溶解试验。[14]King MJ, Garçon L, Hoyer JD, et al; International Council for Standardization in Haematology (ICSH). ICSH guidelines for the laboratory diagnosis of nonimmune hereditary red cell membrane disorders. Int J Lab Hematol. 2015;37:304-325.http://onlinelibrary.wiley.com/doi/10.1111/ijlh.12335/fullhttp://www.ncbi.nlm.nih.gov/pubmed/25790109?tool=bestpractice.com 它使用 20 μL 全血,并在加入甘油之前和之后使用红细胞悬浮液测量吸光度降至其原始值的一半所花费的时间。该试验显示,识别 HS 的敏感性为 98.3%,特异性为 91.1%,[21]Hoffmann JJ, Swaak-Lammers N, Breed WP, et al. Diagnostic utility of the pre-incubated acidified glycerol lysis test in haemolytic and non-haemolytic anaemias. Eur J Haematol. 1991;47:367-370.http://www.ncbi.nlm.nih.gov/pubmed/1761123?tool=bestpractice.com 但在 AIHA、妊娠、骨髓增生异常和其他一些疾病患者中也呈阳性。它也没有被广泛使用。
传统的做法是将渗透脆性 (osmotic fragility, OF) 试验作为 HS 的确认试验。此试验检测与正常红细胞相比,在氯化钠浓度梯度下球形红细胞对溶解的敏感性增加。尽管通过将血液在 37℃(98.6°F)温育 24 小时来提高敏感性,但该试验不具有特异性,在存在球形红细胞的任何疾病(例如 AIHA)时,渗透脆性均增加。[14]King MJ, Garçon L, Hoyer JD, et al; International Council for Standardization in Haematology (ICSH). ICSH guidelines for the laboratory diagnosis of nonimmune hereditary red cell membrane disorders. Int J Lab Hematol. 2015;37:304-325.http://onlinelibrary.wiley.com/doi/10.1111/ijlh.12335/fullhttp://www.ncbi.nlm.nih.gov/pubmed/25790109?tool=bestpractice.com 它很耗时,并且可能在婴儿和轻度疾病病例中呈假阴性结果。[5]Bolton-Maggs PH, Langer JC, Iolascon A, et al; General Haematology Task Force of the British Committee for Standards in Haematology. Guidelines for the diagnosis and management of hereditary spherocytosis - 2011 update. Br J Haematol. 2012;156:37-49.http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2141.2011.08921.x/fullhttp://www.ncbi.nlm.nih.gov/pubmed/22055020?tool=bestpractice.com 该试验会遗漏约 20% 的轻度 HS 病例,因此不再推荐使用。[22]Korones D, Pearson HA. Normal erythrocyte osmotic fragility in hereditary spherocytosis. J Pediatr. 1989;114:264-266.http://www.ncbi.nlm.nih.gov/pubmed/2915286?tool=bestpractice.com
对疑难病例的附加试验
对于 HS 的典型病例,不需要任何附加试验。在以下情况下,需要进行附加试验:
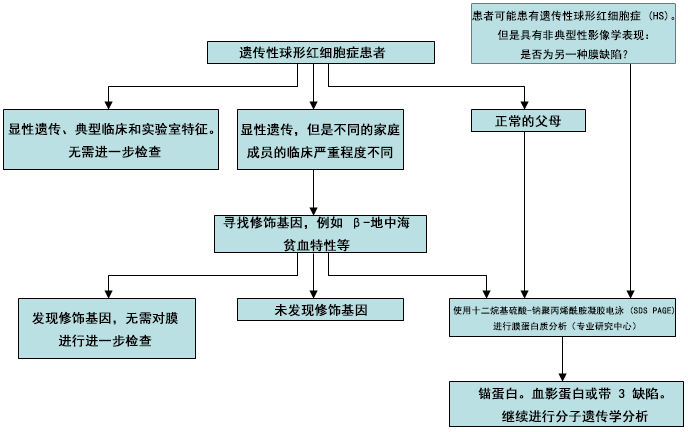
排除不能进行脾切除术的罕见红细胞疾病类型十分重要。[5]Bolton-Maggs PH, Langer JC, Iolascon A, et al; General Haematology Task Force of the British Committee for Standards in Haematology. Guidelines for the diagnosis and management of hereditary spherocytosis - 2011 update. Br J Haematol. 2012;156:37-49.http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2141.2011.08921.x/fullhttp://www.ncbi.nlm.nih.gov/pubmed/22055020?tool=bestpractice.com 对于可能与 HS 混淆的先天性红细胞生成异常性贫血 II 型 (congenital dyserythropoietic anaemia type II, CDA-II) ,脾切除术几乎没有价值。对于这些非典型病例,可以通过十二烷基硫酸钠-聚丙烯酰胺凝胶电泳 (sodium dodecyl sulfate-polyacrylamide gel electrophoresis, SDS-PAGE) 进行定量蛋白质分析(但不是广泛可用的)。SDS-PAGE 分析红细胞膜含量,并通过显示血影蛋白、锚蛋白、带 3 蛋白和蛋白质 4.2(在 HS 中可能受到影响的膜蛋白)的蛋白质条带来确定哪种膜蛋白有缺陷。[14]King MJ, Garçon L, Hoyer JD, et al; International Council for Standardization in Haematology (ICSH). ICSH guidelines for the laboratory diagnosis of nonimmune hereditary red cell membrane disorders. Int J Lab Hematol. 2015;37:304-325.http://onlinelibrary.wiley.com/doi/10.1111/ijlh.12335/fullhttp://www.ncbi.nlm.nih.gov/pubmed/25790109?tool=bestpractice.com 一项针对意大利 300 名 HS 患者的调查显示,带 3 蛋白和血影蛋白缺陷是最常见的蛋白质异常。[3]Mariani M, Barcellini W, Vercellati C, et al. Clinical and hematologic features of 300 patients affected by hereditary spherocytosis grouped according to the type of the membrane protein defect. Haematologica. 2008;93:1310-1317.http://www.haematologica.org/content/93/9/1310.fullhttp://www.ncbi.nlm.nih.gov/pubmed/18641031?tool=bestpractice.com
遗传学分析
虽然已经在 5 种细胞骨架蛋白(α- 和 β-血影蛋白、锚蛋白、带 3 蛋白和蛋白质 4.2)中发现了突变,[4]Iolascon A, Avvisati RA. Genotype/phenotype correlation in hereditary spherocytosis. Haematologica. 2008;93:1283-1288.http://www.haematologica.org/content/93/9/1283.fullhttp://www.ncbi.nlm.nih.gov/pubmed/18757847?tool=bestpractice.com 但在以前的遗传分析是一种处于研究中的检查,尚未普遍可用或有必要使用。[15]Bolton-Maggs PH, King MJ. Hereditary spherocytosis, hereditary elliptocytosis and related disorders. In: Young NS, Gershon SL, High KA, eds. Clinical hematology. Philadelphia, PA: Mosby Elsevier; 2006:293-307.[23]Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372:1411-1426.http://www.ncbi.nlm.nih.gov/pubmed/18940465?tool=bestpractice.com 然而,随着下一代测序的发展,遗传分析将变得更加容易。多数显性 HS 病例是由锚蛋白、带 3 蛋白或 β-血影蛋白的基因突变引起的。这些基因突变可能导致其他骨架蛋白的继发性缺陷。蛋白质 4.2 的基因突变在日本更为常见。非显性遗传性 HS 的发生可能是由于遗传亲本一方的致病突变和另一方的沉默低表达的等位基因,或者可能由新生突变导致。
