家庭医师和肾脏科医师比其他专家更可能作出诊断。常染色体显性遗传性 PKD (ADPKD) 家族史阳性,伴随肾脏体征和症状和/或肾外表现,高度提示 ADPKD。然而,明确诊断需要肾脏的影像学检查或者基因检测(如果影像学检查不能确诊)。对于无阳性家族史的患者,可以考虑推定诊断,无其他表现、存在伴或不伴肝囊肿的肾囊肿提示其他肾囊性疾病。然而这些患者需要接受基因检测以确诊。大约 5% 无家族史的患者会有新突变。[40]Ravine D, Gibson RN, Walker RG, et al. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet. 1994 Apr 2;343(8901):824-7.http://www.ncbi.nlm.nih.gov/pubmed/7908078?tool=bestpractice.com[41]Nicolau C, Torra R, Badenas C, et al. Autosomal dominant polycystic kidney disease types 1 and 2: assessment of US sensitivity for diagnosis. Radiology. 1999 Oct;213(1):273-6.http://www.ncbi.nlm.nih.gov/pubmed/10540671?tool=bestpractice.com
病史
家族史可包括 ADPKD、ESRD、颅内动脉瘤、出血性卒中或蛛网膜下腔出血。常见的主要症状包括胁腹痛/腹痛、肾绞痛和肉眼血尿,少数情况下还有头痛。[6]Iglesias CG, Torres VE, Offord KP, et al. Epidemiology of adult polycystic kidney disease, Olmsted County, Minnesota: 1935-1980. Am J Kidney Dis. 1983 May;2(6):630-9.http://www.ncbi.nlm.nih.gov/pubmed/6846334?tool=bestpractice.com[7]Dalgaard OZ. Bilateral polycystic disease of the kidneys: a follow-up of two hundred and eighty-four patients and their families. Acta Med Scand Suppl. 1957;328:1-255.http://www.ncbi.nlm.nih.gov/pubmed/13469269?tool=bestpractice.com[8]Gabow PA. Autosomal dominant polycystic kidney disease. N Engl J Med. 1993 Jul 29;329(5):332-42.http://www.ncbi.nlm.nih.gov/pubmed/8321262?tool=bestpractice.com 在某一时刻,囊肿见于 50%-75% 的 PKD 患者。[40]Ravine D, Gibson RN, Walker RG, et al. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet. 1994 Apr 2;343(8901):824-7.http://www.ncbi.nlm.nih.gov/pubmed/7908078?tool=bestpractice.com 患者通常出现排尿困难、尿急和耻骨弓上疼痛病史。累及肾实质或囊肿的 UTI 通常表现为胁腹痛和发热。肾结石患者可能出现胁腹痛、血尿、排尿困难和发热。重症肝病患者可能出现胃灼热、反流、恶心、呼吸困难、早饱或腹围增加。
体格检查
腹部检查经常可触及肾脏或肝脏肿块。[6]Iglesias CG, Torres VE, Offord KP, et al. Epidemiology of adult polycystic kidney disease, Olmsted County, Minnesota: 1935-1980. Am J Kidney Dis. 1983 May;2(6):630-9.http://www.ncbi.nlm.nih.gov/pubmed/6846334?tool=bestpractice.com[7]Dalgaard OZ. Bilateral polycystic disease of the kidneys: a follow-up of two hundred and eighty-four patients and their families. Acta Med Scand Suppl. 1957;328:1-255.http://www.ncbi.nlm.nih.gov/pubmed/13469269?tool=bestpractice.com[8]Gabow PA. Autosomal dominant polycystic kidney disease. N Engl J Med. 1993 Jul 29;329(5):332-42.http://www.ncbi.nlm.nih.gov/pubmed/8321262?tool=bestpractice.com 高血压是常见症状,且经常发生在相对年轻的年龄。对于 20 至 34 岁的患者,在任何其他临床表现之前检测出高血压经常是首次发现该疾病的方式。可能还有肾功能不全/ESRD 的体征或症状。经常出现腹股沟、切口或脐旁疝以及腹直肌分离。可能出现心脏杂音,提示二尖瓣脱垂、二尖瓣反流、主动脉瓣反流或主动脉根部扩张。
实验室检查
首先应进行血清电解质、血尿素、肌酐和空腹血脂检查。肌酐可用于评估 GFR。升高的血脂可能与进行性肾功能不全有更大的关联。
应对所有患者进行尿液分析,以检测尿白蛋白排泄是否增加或是否存在蛋白尿。如果发现尿白蛋白排泄增加或蛋白尿,则表明进展至慢性肾脏病的可能性更高。ADPKD 患者中尿白蛋白排泄增加还与 LVH 的高发率有关。显微镜下或肉眼血尿常见。这些患者中可能出现白细胞尿,但这并不总是表明 UTI,这种情况下应进行尿培养。如果有 UTI 症状或发热症状,应在最初评估时进行尿培养。即使有严重的尿路感染,尿培养仍有可能显示阴性,因为囊肿并不与尿道相通。对于所有代谢活跃的结石病患者,应收集 24 小时尿液以进行尿液生化检查(尿量、pH 值、草酸、尿酸、柠檬酸盐、磷酸盐、钠和钙,以及肌酐以评估收集的完整性),并计算过饱和度。
肾脏影像
影像显示肾囊肿伴/或不伴肝囊肿。成像的益处包括:证实可能影响生育计划的诊断,早期识别和治疗疾病并发症,以及选择基因上不受影响的家庭成员进行活体捐赠移植。在检查前,应给予恰当的辅导。[42]Kielstein R, Sass HM. Genetics in kidney disease: how much do we want to know? Am J Kidney Dis. 2002 Mar;39(3):637-52.http://www.ncbi.nlm.nih.gov/pubmed/11877585?tool=bestpractice.com 应讨论在保险和就业方面与阳性诊断相关的可能歧视。
由于低成本和安全性,超声检查是最常见的初次检查。具有 50% 疾病风险患者的超声诊断标准包括:30 岁以下患者至少 2 处单侧或双侧囊肿;30 至 59 岁患者每个肾脏 2 处囊肿;60 岁及以上患者每个肾脏 4 处囊肿。[40]Ravine D, Gibson RN, Walker RG, et al. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet. 1994 Apr 2;343(8901):824-7.http://www.ncbi.nlm.nih.gov/pubmed/7908078?tool=bestpractice.com 对于 30 岁及以上患者或更年轻的 PKD1 突变患者,这些标准的敏感性几乎是 100%,但对于 30 岁以下 PKD2 突变的患者,敏感性只有 67%。[40]Ravine D, Gibson RN, Walker RG, et al. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet. 1994 Apr 2;343(8901):824-7.http://www.ncbi.nlm.nih.gov/pubmed/7908078?tool=bestpractice.com[41]Nicolau C, Torra R, Badenas C, et al. Autosomal dominant polycystic kidney disease types 1 and 2: assessment of US sensitivity for diagnosis. Radiology. 1999 Oct;213(1):273-6.http://www.ncbi.nlm.nih.gov/pubmed/10540671?tool=bestpractice.com 对后一组患者,应使用 CT 扫描或 MRI。在具有 50% 疾病风险的儿童中,大的回声肾(无明显肉眼囊肿)是诊断症状。已经开发了诊断 ADPKD 高危人群(家庭成员患有 ADPKD)的新超声诊断标准。[43]Pei Y, Obaji J, Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009 Jan;20(1):205-12.http://jasn.asnjournals.org/content/20/1/205.fullhttp://www.ncbi.nlm.nih.gov/pubmed/18945943?tool=bestpractice.com 这些标准不适用于 MRI 或 CT 扫描,因为这可能造成假阳性结果。
如果超声扫描不能确诊,特别是对 30 岁以下的 PKD2 突变患者,应使用造影剂增强的 CT 扫描或 MRI。因为 CT 和 MRI 比超声检查更敏感,上述超声标准并不适用于这些技术。需要权衡 CT 使用与辐射剂量。如果每个肾有 10 处以上囊肿,尽管无家族史,没有可以解释表型的其他囊性肾脏病的肾脏或肾外表现证据,那么可以考虑 ADPKD 的假定诊断。在初始评估时,对于肾功能正常的患者,CT 扫描(增强和无增强)可以提供关于肾脏囊肿受累严重程度和肾实质相对保存的有用信息。这将有助于判断预后以及肾功能下降的可能性。  [Figure caption and citation for the preceding image starts]: 轻度疾病患者的腹部和肝脏 CT 扫描来自 Dr M. Hogan 的收藏 [Citation ends].
[Figure caption and citation for the preceding image starts]: 轻度疾病患者的腹部和肝脏 CT 扫描来自 Dr M. Hogan 的收藏 [Citation ends].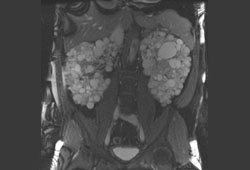 [Figure caption and citation for the preceding image starts]: 症状性多囊肾病患者的腹部和盆腔 MRI来自 Dr M. Hogan 的收藏 [Citation ends].
[Figure caption and citation for the preceding image starts]: 症状性多囊肾病患者的腹部和盆腔 MRI来自 Dr M. Hogan 的收藏 [Citation ends].
根据 CT 扫描的肾脏总体积,基于影像的分类系统可用于识别疾病进展更快速的潜在病例。该分类系统可改善选择患者纳入临床试验。[44]Irazabal MV, Rangel LJ, Bergstralh EJ, et al. Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials. J Am Soc Nephrol. 2015 Jan;26(1):160-72.http://www.ncbi.nlm.nih.gov/pubmed/24904092?tool=bestpractice.com
肾外检查
CT 扫描也可提供肾外囊肿的证据。肝囊肿是最常见的肾外表现。胰腺囊肿(患病率 9%)几乎总是无症状的,伴随罕见的复发性胰腺炎。它们不太可能与管内乳头状黏液性肿瘤或癌有关。此外,将识别出肾结石,并区分结石病与囊壁钙化或实质钙化。大约 20% 的患者有肾结石,其成分通常为尿酸或草酸钙。[45]Torres VE, Erickson SB, Smith LH, et al. The association of nephrolithiasis and autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1988 Apr;11(4):318-25.http://www.ncbi.nlm.nih.gov/pubmed/3354568?tool=bestpractice.com[46]Torres VE, Wilson DM, Hattery RR, et al. Renal stone disease in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1993 Oct;22(4):513-9.http://www.ncbi.nlm.nih.gov/pubmed/8213789?tool=bestpractice.com[47]Grampsas SA, Chandhoke PS, Fan J, et al. Anatomic and metabolic risk factors for nephrolithiasis in patients with autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2000 Jul;36(1):53-7.http://www.ncbi.nlm.nih.gov/pubmed/10873872?tool=bestpractice.com 在 ADPKD 患者中,低尿柠檬酸盐水平和尿液 PH 值是诱发结石形成的主要代谢因素。[45]Torres VE, Erickson SB, Smith LH, et al. The association of nephrolithiasis and autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1988 Apr;11(4):318-25.http://www.ncbi.nlm.nih.gov/pubmed/3354568?tool=bestpractice.com 可以考虑使用 KUB 和 X 线断层成像区分尿酸结石(可透射线)和钙结石(不透射线)。[46]Torres VE, Wilson DM, Hattery RR, et al. Renal stone disease in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1993 Oct;22(4):513-9.http://www.ncbi.nlm.nih.gov/pubmed/8213789?tool=bestpractice.com 双能 CT 是另一种鉴别尿酸结石与其他(非尿酸)肾结石的有效方法。[46]Torres VE, Wilson DM, Hattery RR, et al. Renal stone disease in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1993 Oct;22(4):513-9.http://www.ncbi.nlm.nih.gov/pubmed/8213789?tool=bestpractice.com
确定先兆性头痛和及时诊断及治疗漏动脉瘤是预后的重要决定因素。在检测急性颅内出血时,脑部薄层非增强 CT 扫描比脑部 MRI 更敏感。大多数微动脉瘤往往是在脑前循环,并且较小。如果 CT 是阴性但可疑指数仍然很高,应进行腰椎穿刺术或磁共振血管造影检查。如果有颅内出血,腰椎穿刺术将显示脑脊液中的红细胞和皮肤黄染(在脑脊液离心后见到的黄色或粉红色调),这是由于血红蛋白的降解产物造成的。如果这些检查阳性,患者应立即进入神经外科 ICU 进行常规的血管造影和漏血性颅内动脉瘤治疗。
 成人诊断性腰椎穿刺的动画演示
成人诊断性腰椎穿刺的动画演示
对于心脏杂音或疑似左心室功能障碍患者,应进行心电图检查。对于特定的存在心脏杂音的患者,可考虑进行超声心动图检查,以确定双心室舒张功能紊乱和主动脉根部扩张。
基因检测
基因检测可用于以下病例:
成像结果是模棱两可或不确定的。
在没有家族病史的情况下确定诊断(这些患者的最后诊断取决于突变分析)。
当年轻患者需要明确的诊断时,例如潜在的活体肾脏捐赠者。
很少考虑针对 ADPKD 进行出生前和胚胎植入前的基因检测。[54]Sujansky E, Kreutzer SB, Johnson AM, et al. Attitudes of at-risk and affected individuals regarding presymptomatic testing for autosomal dominant polycystic kidney disease. Am J Med Genet. 1990 Apr;35(4):510-5.http://www.ncbi.nlm.nih.gov/pubmed/2333880?tool=bestpractice.com[55]De Rycke M, Georgiou I, Sermon K, et al. PGD for autosomal dominant polycystic kidney disease type 1. Mol Hum Reprod. 2005 Jan;11(1):65-71.https://academic.oup.com/molehr/article/11/1/65/987966http://www.ncbi.nlm.nih.gov/pubmed/15591452?tool=bestpractice.com 体外受精后胚胎植入前遗传学诊断 (preimplantation genetic diagnosis, PGD) 并非常规可用。[56]Tuffs A. Court allows preimplantation genetic diagnosis in Germany. BMJ. 2010 Jul 12;341:c3741.http://www.ncbi.nlm.nih.gov/pubmed/20624828?tool=bestpractice.com
大约 5% 的阴性家族史患者会有新突变。[40]Ravine D, Gibson RN, Walker RG, et al. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet. 1994 Apr 2;343(8901):824-7.http://www.ncbi.nlm.nih.gov/pubmed/7908078?tool=bestpractice.com[41]Nicolau C, Torra R, Badenas C, et al. Autosomal dominant polycystic kidney disease types 1 and 2: assessment of US sensitivity for diagnosis. Radiology. 1999 Oct;213(1):273-6.http://www.ncbi.nlm.nih.gov/pubmed/10540671?tool=bestpractice.com PKD1 基因突变比 PKD2 基因突变导致的疾病更严重。[57]Gabow PA, Johnson AM, Kaehny WD, et al. Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease. Kidney Int. 1992 May;41(5):1311-9.http://www.ncbi.nlm.nih.gov/pubmed/1614046?tool=bestpractice.com 可以通过连锁分析或序列分析完成基因检测,但是序列分析是首选。对于 ADPKD 患者,使用完整的基因测序筛查可检出高达 91% 病例的突变,65% 有截短突变,这可以很容易地用于诊断。[58]Rossetti S, Consugar MB, Chapman AB, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2007 Jul;18(7):2143-60.http://jasn.asnjournals.org/content/18/7/2143.fullhttp://www.ncbi.nlm.nih.gov/pubmed/17582161?tool=bestpractice.com 在 ADPKD 突变数据库中,可找到所有发表的突变及其他变异。PKD Foundation: ADPKD mutation database 然而,突变分析成本昂贵,而且通常不是必需的。[58]Rossetti S, Consugar MB, Chapman AB, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2007 Jul;18(7):2143-60.http://jasn.asnjournals.org/content/18/7/2143.fullhttp://www.ncbi.nlm.nih.gov/pubmed/17582161?tool=bestpractice.com
如果知道一位家庭成员有突变,可以在其他家庭成员中以相对低得多的成本确认这个突变。PKD1 和 PKD2 基因测序在某些情况下很有帮助:30 岁以下的活体亲属潜在供者,或者高度怀疑 PKD2 时;以及在阴性家族史患者(或活体亲缘供体)中,影像学检查无法诊断时。[59]Hannig VL, Erickson SM, Phillips JA 3rd. Utilization and evaluation of living-related donors for patients with adult polycystic kidney disease. Am J Med Genet. 1992 Nov 1;44(4):409-12.http://www.ncbi.nlm.nih.gov/pubmed/1442877?tool=bestpractice.com[60]Connor A, Lunt PW, Dolling C, et al. Mosaicism in autosomal dominant polycystic kidney disease revealed by genetic testing to enable living related renal transplantation. Am J Transplant. 2008 Jan;8(1):232-7.http://www.ncbi.nlm.nih.gov/pubmed/17973957?tool=bestpractice.com PKD1 体积大、复杂,以及有明显的等位基因异质性是直接采用 DNA 分析进行分子检测的阻碍。
连锁分析使用了高度信息化的微卫星标记侧腹 PKD1 和 PKD2,并且要求诊断精确、有关家庭成员可用和足够且愿意接受检测。因为这些限制,连锁分析只适于不足 50% 的家庭。连锁分析经常是不可行的,并且检出率<100%。在具有连锁特征的人群中,PKD1 占了病例的大约 85%,余下的大部分是 PKD2。现已很少使用连锁分析,已经被完全基因测序代替。
另一个新兴的方法可能是全外显子组测序,但是该方法的敏感性和特异性在 ADPKD 人群中并不十分明确,并且由于同源 PKD1 基因的存在,该方法较为复杂。
 静脉穿刺和抽血的动画演示
静脉穿刺和抽血的动画演示
 关于如何进行心电图的动画演示
关于如何进行心电图的动画演示
